
Avogadro stands as a premier open-source molecular editor and visualization tool, specifically engineered to cater to the exacting demands of scientists, researchers, and students across various scientific disciplines. This powerful application provides a comprehensive platform for the intuitive manipulation and detailed rendering of three-dimensional molecular structures, allowing users to explore chemical compositions from every conceivable angle and perspective. Its cross-platform compatibility ensures accessibility for a broad user base, making it an indispensable asset in fields ranging from computational chemistry and molecular modeling to bioinformatics and materials science.
The fundamental objective behind Avogadro’s design is to democratize 3D molecular manipulation, transforming what could be a complex task into an easily manageable and even engaging experience. Even for those without a deep scientific background, the software offers a surprising degree of satisfaction in simply crafting and visualizing custom-colored molecular structures. This ease of use is primarily achieved through a user-friendly interface that allows for direct manipulation of molecules using standard mouse controls, enabling users to effortlessly drag and rotate structures along their three axes. Beyond its immediate utility as a visual editor, Avogadro distinguishes itself through a robust and flexible architecture that supports extensive customization and development via plugins, promising even greater versatility with future scripting options. While its open-source nature, characteristic of collaborative projects, may sometimes entail encounters with bugs or occasional crashes, its overall value as a sophisticated scientific instrument, combined with its capacity for user-driven development, solidifies its position as a cornerstone tool in chemical and material sciences.
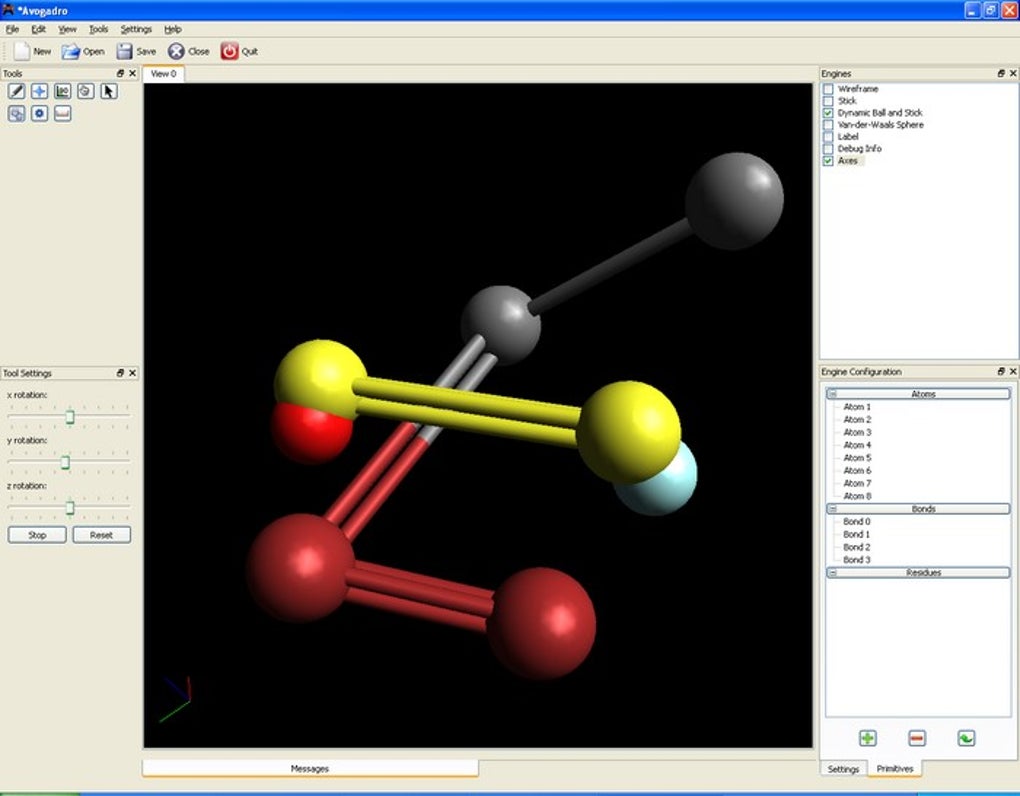
Key Features and Intuitive 3D Molecular Manipulation
At its core, Avogadro is celebrated for its highly intuitive and powerful 3D molecular structure editing capabilities. The software empowers users to construct, modify, and visualize complex molecular geometries with remarkable ease, making it a cornerstone tool for both educational purposes and advanced research. The process of “drawing” or building three-dimensional molecular compositions within Avogadro is designed to be as straightforward as possible, simulating a virtual laboratory bench where atoms can be placed, bonded, and arranged to form intricate molecules.
Users begin by selecting specific elements from the periodic table, which can then be placed within the 3D workspace. Avogadro intelligently handles bonding, automatically suggesting bond types (single, double, triple) and geometries based on the selected atoms and their common valencies. This intelligent assistance accelerates the building process, allowing researchers to quickly assemble even large and complex structures like proteins or polymers. Beyond mere construction, the software provides a comprehensive suite of tools for modifying existing structures. Atoms can be easily repositioned, deleted, or replaced, and bond orders can be adjusted with a few clicks. This level of granular control is crucial for refining molecular models, optimizing geometries, and exploring conformational changes.
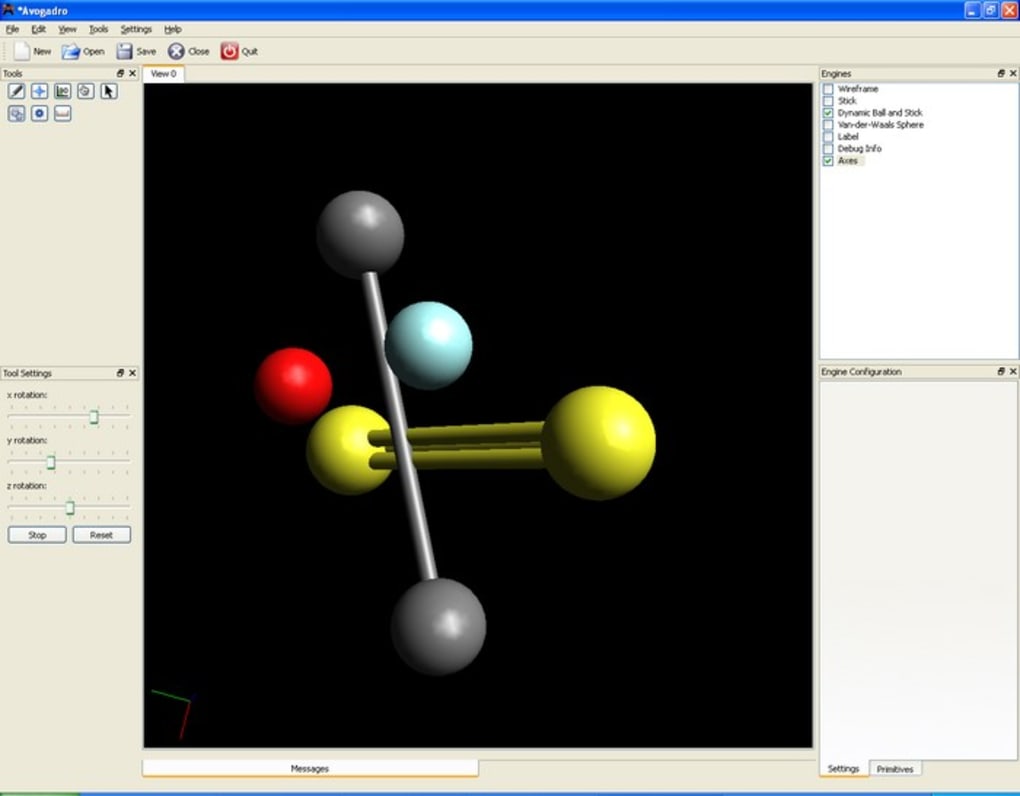
One of Avogadro’s most compelling features is its dynamic visualization engine. Once a molecule is built, users can interactively rotate, translate, and zoom in on the structure, viewing it from any conceivable angle and perspective. This 360-degree observational capability is vital for understanding spatial relationships between atoms, identifying steric clashes, and appreciating the intricate three-dimensional architecture of molecules. The software also offers various rendering styles, from simple wireframes to ball-and-stick, space-filling, and even more advanced representations that highlight molecular orbitals or electrostatic potential surfaces. These visual options are not merely aesthetic; they serve crucial analytical purposes, helping scientists to discern specific features, analyze interactions, and present their findings with clarity. The ability to color-code atoms, bonds, and other molecular properties further enhances this visual clarity, allowing for custom highlighting of functional groups, charge distributions, or specific structural motifs.
Furthermore, Avogadro goes beyond static structure editing by offering tools for geometry optimization and vibrational analysis. Integrating with external computational chemistry packages, Avogadro can act as a pre- and post-processor, preparing input files for quantum chemistry calculations and then visualizing the results. This seamless workflow is invaluable for computational chemists who need to predict molecular properties, reaction pathways, or spectroscopic data. The software’s flexible rendering framework is a key differentiator, providing high-quality graphics that are essential for both in-depth analysis and compelling presentations. Whether it’s for illustrating a concept in a lecture, analyzing experimental data, or developing new materials, Avogadro’s intuitive 3D manipulation tools and sophisticated rendering capabilities make it an indispensable asset for anyone working with molecular structures.
Broad Applications Across Scientific Disciplines
Avogadro’s versatile design makes it an invaluable tool across a multitude of scientific disciplines, serving both academic instruction and cutting-edge research. Its capacity for intuitive 3D molecular manipulation translates directly into practical applications in areas such as computational chemistry, molecular modeling, bioinformatics, and materials science. Each of these fields benefits uniquely from Avogadro’s ability to visualize, construct, and analyze molecular structures in a three-dimensional context.
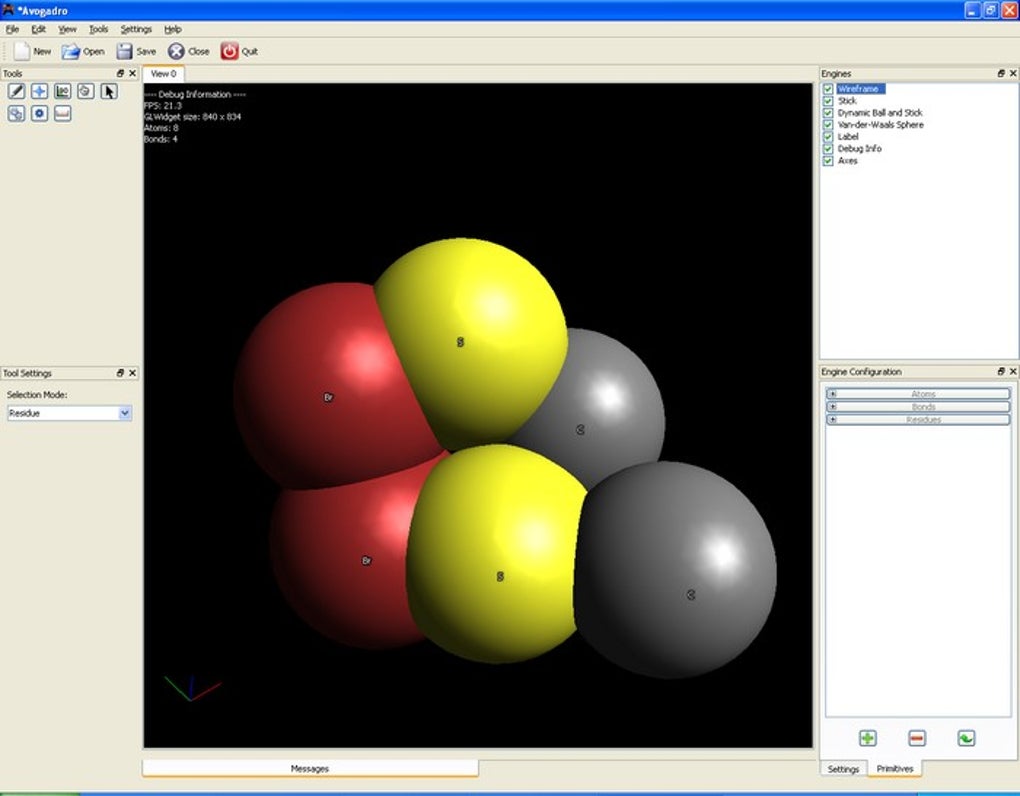
In computational chemistry, Avogadro often serves as the initial gateway for researchers. Before running computationally intensive simulations (e.g., density functional theory, molecular dynamics), scientists need to build and optimize initial molecular geometries. Avogadro excels here by providing an easy-to-use graphical interface to construct molecules, adjust bond lengths and angles, and even set up periodic boundary conditions for solid-state simulations. It can generate input files for a wide array of popular quantum chemistry and molecular mechanics software packages, significantly streamlining the pre-processing workflow. Post-calculation, Avogadro becomes a powerful visualization tool, allowing chemists to view optimized structures, molecular orbitals, electron density maps, vibrational modes, and reaction pathways. This visual feedback is crucial for interpreting complex data and validating theoretical predictions.
Molecular modeling broadly encompasses the theoretical methods used to model the behavior of molecules. Avogadro’s role here is foundational. It allows students and researchers to build models of small organic molecules, inorganic complexes, or even larger biological macromolecules like peptides and oligonucleotides. By visually exploring different conformations and interactions, users can develop a deeper understanding of molecular properties, drug-receptor binding, or catalytic mechanisms. Its 3D environment helps in conceptualizing spatial relationships that are difficult to grasp from 2D diagrams alone, fostering a more intuitive understanding of molecular architecture and dynamics.
In bioinformatics, particularly structural bioinformatics, Avogadro provides a simplified approach to visualizing the building blocks of life. While specialized tools exist for large protein and DNA structures, Avogadro is excellent for working with smaller ligands, active site residues, or specific motifs within larger biomolecules. It can be used to prepare models of drug candidates, analyze their interactions with target proteins, or visualize mutations. For students, it’s an accessible way to understand the chemistry behind biological processes, offering a visual bridge between chemical structure and biological function.
Materials science is another major beneficiary of Avogadro. Researchers in this field often design new materials with specific properties, whether it be catalysts, polymers, semiconductors, or nanomaterials. Avogadro allows them to construct unit cells for crystalline structures, build polymer chains, or design nanoparticles. The ability to visualize these structures in 3D helps in understanding how atomic arrangements influence bulk material properties. For instance, simulating the arrangement of atoms in a crystal lattice or observing the interconnections in a metal-organic framework (MOF) becomes a tangible process. This visualization is critical for predicting mechanical strength, electronic band gaps, or adsorption capacities of designed materials.
Beyond these specific fields, Avogadro is also widely adopted in education. Its user-friendly interface makes it an excellent teaching aid for chemistry, physics, and biology courses at all levels. Students can build molecules, experiment with different isomers, observe molecular symmetry, and understand concepts like hybridization or stereochemistry in an interactive 3D environment. This hands-on experience deepens learning and makes abstract concepts more concrete.
In essence, Avogadro serves as a versatile digital workbench that bridges the gap between theoretical concepts and tangible visualizations across numerous scientific disciplines. Its ease of use, combined with its powerful features, makes it an indispensable tool for anyone seeking to explore, understand, and manipulate the molecular world.
Empowering Customization: Plugin Architecture and Scripting
One of the most significant strengths of Avogadro, setting it apart in the realm of scientific software, is its highly flexible and extensible nature, primarily driven by a powerful plugin architecture and the promise of future scripting options. This design philosophy directly addresses the diverse and evolving needs of the scientific community, allowing users to tailor the software to specific research workflows and integrate it seamlessly with other tools.
The concept of plugins allows Avogadro to remain lightweight and efficient while offering a vast array of specialized functionalities. Instead of incorporating every conceivable feature into the core application, which would make it bloated and potentially slow, Avogadro provides a robust framework for external modules – plugins – to extend its capabilities. For users with some coding knowledge, this means they can develop custom plugins to add new types of molecular analysis, implement novel visualization techniques, interface with specialized external databases, or even create entirely new tools for molecular manipulation that are not part of the standard distribution. This open-ended extensibility is invaluable for researchers working on niche problems that might not be covered by off-the-shelf software. For example, a chemist working with a specific type of organometallic complex might develop a plugin to calculate a unique descriptor relevant to their research, or a materials scientist might create a plugin for generating specific crystal defects.
The foundation for much of Avogadro’s chemical intelligence and interoperability lies in its reliance on Open Babel. Open Babel is a peer-reviewed, open-source cheminformatics toolkit, renowned for its ability to interconvert chemical file formats. By being “based on Open Babel,” Avogadro inherits an incredible capacity to read and write a vast number of chemical file formats (e.g., .xyz, .mol, .sdf, .pdb, .cif), making it highly compatible with data generated by virtually any other chemistry software. This deep integration is crucial for research workflows, as scientists rarely use a single program for all their tasks. Avogadro’s ability to seamlessly import structures from computational chemistry packages and export them for further analysis in other tools minimizes data conversion hurdles and maximizes productivity. Furthermore, Open Babel provides a rich library of cheminformatics functionalities, such as calculating molecular properties, performing substructure searches, and generating canonical representations, which plugins can leverage to add sophisticated analytical features to Avogadro.
Looking ahead, the developers have indicated that Avogadro will also offer scripting options. This represents another powerful layer of customization, allowing users to automate repetitive tasks, create complex macros, and integrate Avogadro into larger programmatic workflows without necessarily needing to compile full plugins. Scripting, often done in languages like Python, provides a more accessible entry point for automation and customization, enabling scientists to control the software programmatically for high-throughput analyses or to perform sequential operations that would be tedious to do manually. This feature is particularly exciting for computational researchers who often rely on scripting to manage large datasets and complex simulation setups.
The open-source nature of Avogadro itself is intrinsically linked to this extensibility. As a collaborative project, its development is driven by a community of contributors who share a common goal of creating a powerful and flexible tool. This community model encourages the development and sharing of plugins and scripts, fostering an ecosystem where new functionalities are continually being added and refined. This collective effort ensures that Avogadro remains at the forefront of scientific software, adapting to new challenges and incorporating the latest computational chemistry methodologies. The ability for users to inspect, modify, and contribute to the source code, as well as develop their own extensions, truly empowers the scientific community to shape the tool to their exact specifications, making Avogadro more than just a piece of software – it’s a dynamic platform for scientific innovation.
Navigating the Open-Source Landscape: Stability and Development
Avogadro’s identity as a free, open-source, and cross-platform application brings with it a unique set of advantages and challenges, particularly concerning its stability and ongoing development. While its open-source nature fosters collaboration and innovation, it also means that the software’s journey to maturity can sometimes be characterized by periodic encounters with bugs and errors.
One of the most frequently cited concerns with open-source collaborations, as highlighted in the provided text, is their propensity to be “prone to bugs and errors so it may crash at any time.” This is a reality stemming from the distributed nature of development, where many volunteers contribute code, and comprehensive, centralized quality assurance can sometimes lag behind rapid feature development. Unlike proprietary software developed by dedicated, paid teams with extensive testing resources, open-source projects often rely on community bug reports and peer review. While this process is robust in the long term, it can mean that early or less mature releases might exhibit instability. Users might experience unexpected crashes, rendering glitches, or inconsistencies in functionality, which can be frustrating during critical research tasks.
However, it’s crucial to view this within the context of continuous development. The mention of a specific release marking “the first ‘stable’ release of Avogadro” signifies a critical milestone in the project’s lifecycle. A “stable” release typically indicates that the core functionalities have undergone rigorous testing, major bugs have been addressed, and the developers are confident in its reliability for general use. This transition from more experimental, rapidly evolving versions to a designated stable branch is a testament to the project’s maturation and the community’s commitment to delivering a dependable tool. While stability improves, it doesn’t mean the software becomes entirely bug-free, but rather that the most disruptive issues are typically resolved, and ongoing maintenance focuses on refining performance and fixing minor glitches.
The advantages of the open-source model largely outweigh these initial challenges. Being “open source and free” eliminates financial barriers, making sophisticated molecular modeling accessible to institutions and individuals worldwide who might not have the budget for expensive commercial software. This accessibility fosters broader scientific participation and education. Moreover, the transparency of open-source code means that scientists can inspect exactly how calculations are performed, verify algorithms, and even modify the source code to suit highly specialized needs. This level of control and transparency is often impossible with proprietary software.
Avogadro’s cross-platform nature is another significant benefit derived from its open-source development. It is designed to run seamlessly on Windows, macOS, and Linux operating systems. This flexibility is vital in academic and research environments, where users often work with diverse hardware and operating systems. It ensures that projects can be shared and collaborated on regardless of individual computing preferences, promoting a more integrated research ecosystem.
The community-driven development model also means that issues, when they arise, can often be addressed quickly. Active users report bugs, suggest features, and even contribute code fixes, leading to rapid iteration and improvement. Furthermore, the development “based on Open Babel” ensures that Avogadro benefits from the stability and wide-ranging chemical file format support of another mature open-source project. This integration means Avogadro isn’t reinventing the wheel for fundamental cheminformatics tasks but rather building upon a solid, well-tested foundation.
In conclusion, while the open-source development process can introduce temporary stability challenges, Avogadro’s journey toward “stable” releases, coupled with its inherent benefits of being free, cross-platform, and highly extensible, positions it as a valuable and continuously improving tool. Its success hinges on the active participation of its user and developer community, collectively navigating the open-source landscape to enhance its capabilities and reliability for scientific exploration.
Avogadro has solidified its reputation as an excellent scientific tool, deftly balancing power with user-friendliness. Its core strength lies in its ability to render complex 3D molecular structures in an easily manipulable format, making it accessible not only to seasoned scientists but also to students and even curious individuals who find enjoyment in creating visually appealing chemical compositions. This intuitive interaction, primarily driven by simple mouse controls, significantly lowers the barrier to entry for exploring the intricate world of molecular architecture.
The software’s flexibility is further amplified by its robust plugin architecture. For those with coding expertise, Avogadro transforms into a versatile development platform, allowing for the creation of custom functionalities that can cater to highly specific research needs. This extensibility, combined with the promise of future scripting options, ensures that Avogadro can evolve alongside the ever-changing demands of scientific inquiry. Its foundation in Open Babel guarantees broad compatibility with various chemical file formats, establishing it as a central hub for molecular data exchange across different software ecosystems.
While its open-source nature means that occasional bugs or crashes may occur, a characteristic common to many collaborative development projects, the commitment to “stable” releases signifies a mature and reliable trajectory. The ongoing community contributions and the transparency of its development process continuously refine its performance and expand its capabilities. Available for free across multiple platforms, Avogadro democratizes access to advanced molecular modeling and visualization, making sophisticated scientific tools accessible to a global audience.
In summary, Avogadro stands out as a powerful, adaptable, and community-driven solution for 3D molecular manipulation. Its blend of ease of use, extensive customization options, and broad applicability across computational chemistry, molecular modeling, bioinformatics, and materials science makes it an indispensable asset in both educational settings and advanced research laboratories. Users interested in exploring this vital tool can find more information and download it from PhanMemFree.org.
File Information
- License: “Free”
- Latest update: “July 11, 2023”
- Platform: “Windows”
- OS: “Windows XP”
- Language: “English”
- Downloads: “57.8K”
- Size: “11.29 MB”














